
Step-by-Step Guide to Upload Genomic Data to NGS Cloud
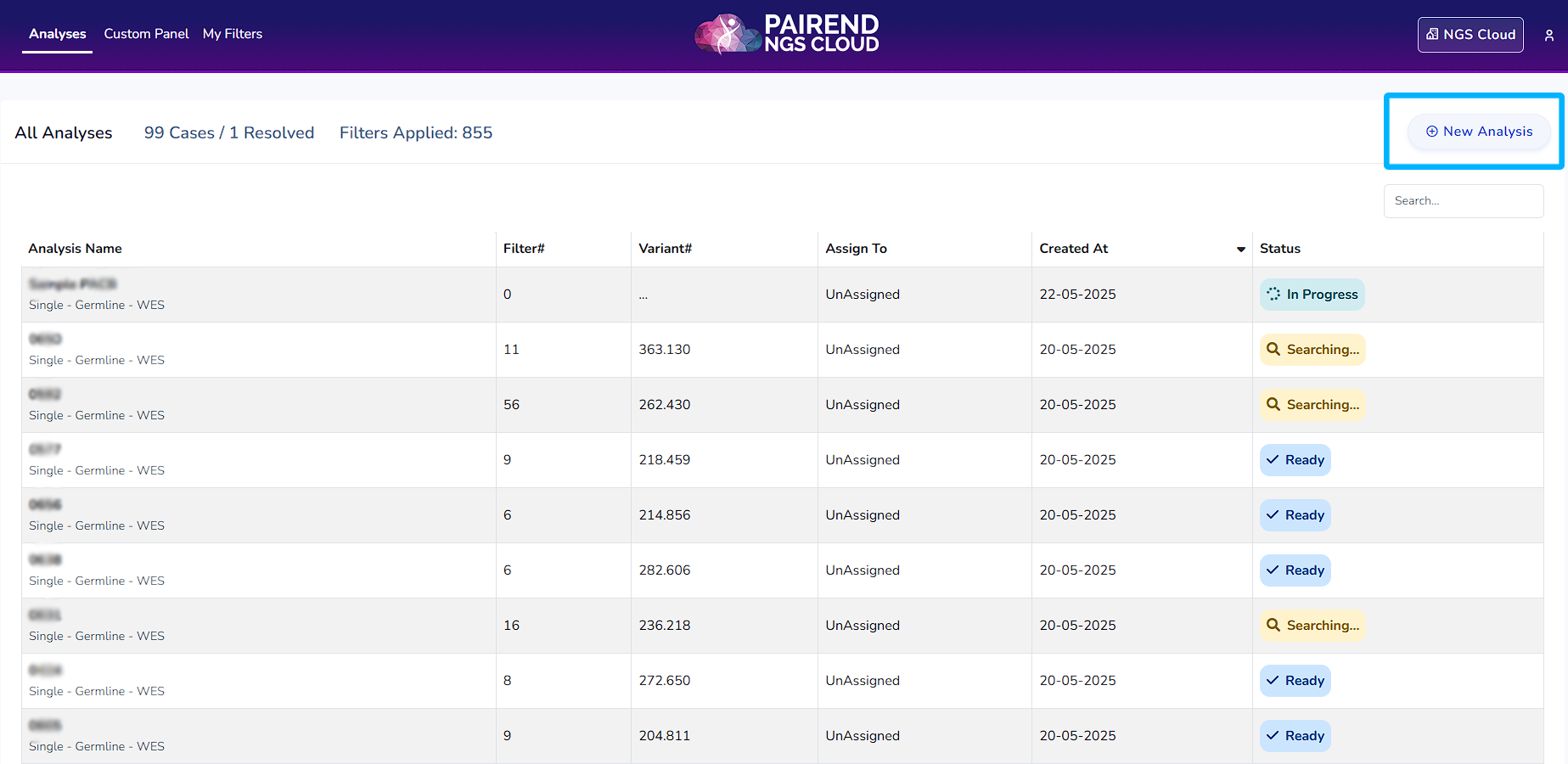
When you log into the NGS Cloud, you will access a dashboard and New Analysis button to upload your data. Click “New Analysis” button to start a new analysis. From the New Analysis section, select the appropriate application for your analysis: Germline, Somatic, Hereditary Cancer, Carrier Risk. You can perform single sample analysis in all applications. You can perform pedigree analysis using data from a family in germline, hereditary cancer and carrier risk analysis. You can also perform cohort analysis in all types of applications.
Selection of Analysis Type

After selecting the application type you will access the area where you can drop files, browse files or import from My device, One Drive, Dropbox, Google Drive and Link. You can upload fastq, bam, cram, vcf files, 100 files, up to 1 TB. We can increase the size limit upon request. Transfer from FTP or from server to server is also possible. Accepted file extensions: fq, .fastq, fq.gz, fastq.gz, .bam, .vcf, vcf.gz
Considerations Regarding the File Names
The filename is split with the underscore (_) character, and the leftmost chunk is assigned to the sample name. Before loading FASTQ files, the read pairs (R1 and R2) must be differentiated for paired-end reads. For a correct classification, it must be specified in _r1 or _r2 (excluding the file extension) in the original file name.
Example 1: The name of your original file => Symbol45_r1.fq.gz
- The sample name on the platform => Symbol45
- Detected file type => Zipped FastQ (FastQ Classification => R1)
Example 2: The name of your original file => Radiguzel_2005_001.vcf.gz
- The sample name on the platform => Radiguzel
- Detected file type => Zipped VCF
After uploading the data, it will appear in the samples section.
To proceed with the analysis, select the data that will be analyzed and enter the analysis information into the Analysis Info section. Genome version used is always hg38. Platform is automatically detected by our system. Capture kit has to be entered as well as the ethnicity, current age of the patient, onset age of the disease and gender. If the marriage is from a consanguinity marriage it also has to be informed. Finally, clinical information has to be entered from ADD Clinic Section
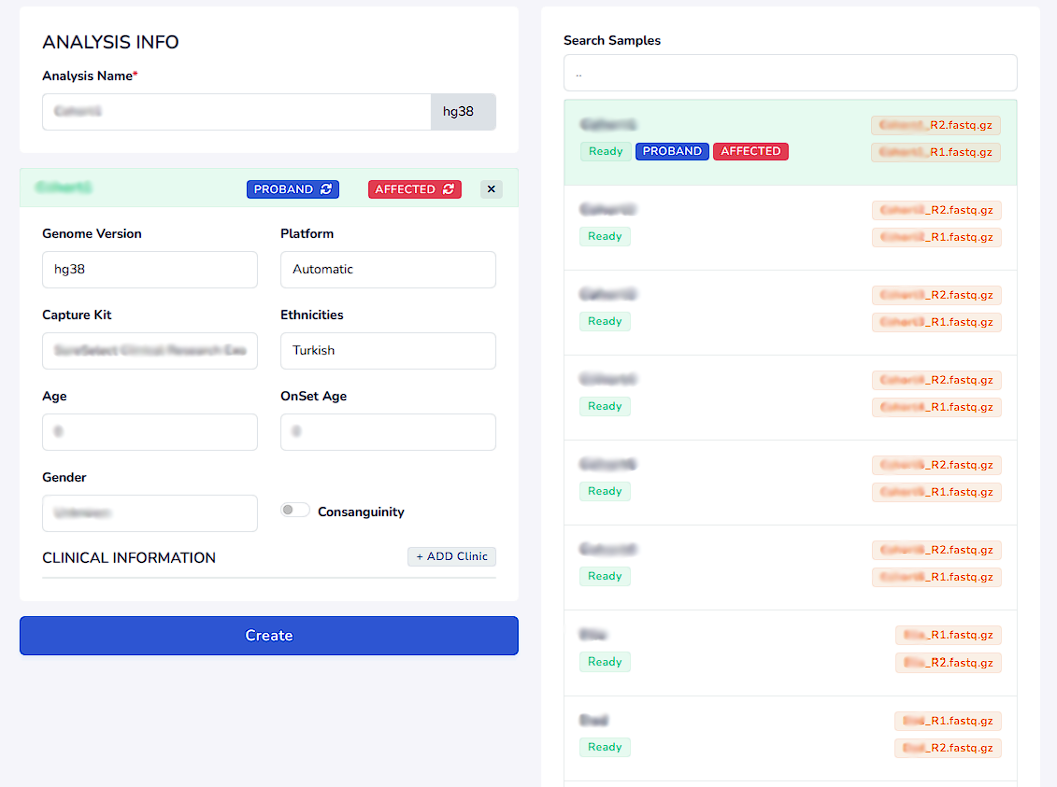
Input Clinical Information
Enter the related genes, phenotypes (HPO terms) and diseases that are related to the case. Press enter after writing the genes, phenotypes and diseases to search for them. A list of related choices will appear as a list within which you can select the clinical information.
Display on Analysis Dashboard
Once you enter the clinical information of the data, it will appear on top in the analysis dashboard. Click Analyses to access the analyses dashboard.
Estimated Processing Times
-
WES Data: 4 to 5 hours
-
WGS Data: 7 to 8 hours
-
VCF Data: 30 minutes to 1 hour
NGS Cloud Analysis Stages
Closed:
The analysis process has been completed and closed by the system. No further changes will occur unless manually restarted.
Diagnosed:
The data has been successfully processed and bioinformatics diagnosis has been completed. Results are available in the system.
Pending Review:
The analysis is complete but awaiting expert or user review before proceeding.
Pending Report:
The diagnosis is complete and the system is waiting for the report to be generated. Data is finalized at this stage.
In Queue:
The analysis request has been received but has not yet started processing. It will be processed in turn.
In Progress:
The analysis is currently running. The system is actively performing bioinformatics computations.
Ready:
The analysis has been completed and results are ready to be accessed. Users can now review or report the findings.
Searching:
Required genomic data, reference sequences, or additional information is being retrieved or loaded by the system.
Creating:
A new analysis session is being initialized. The system is preparing the necessary files and environment for processing.


